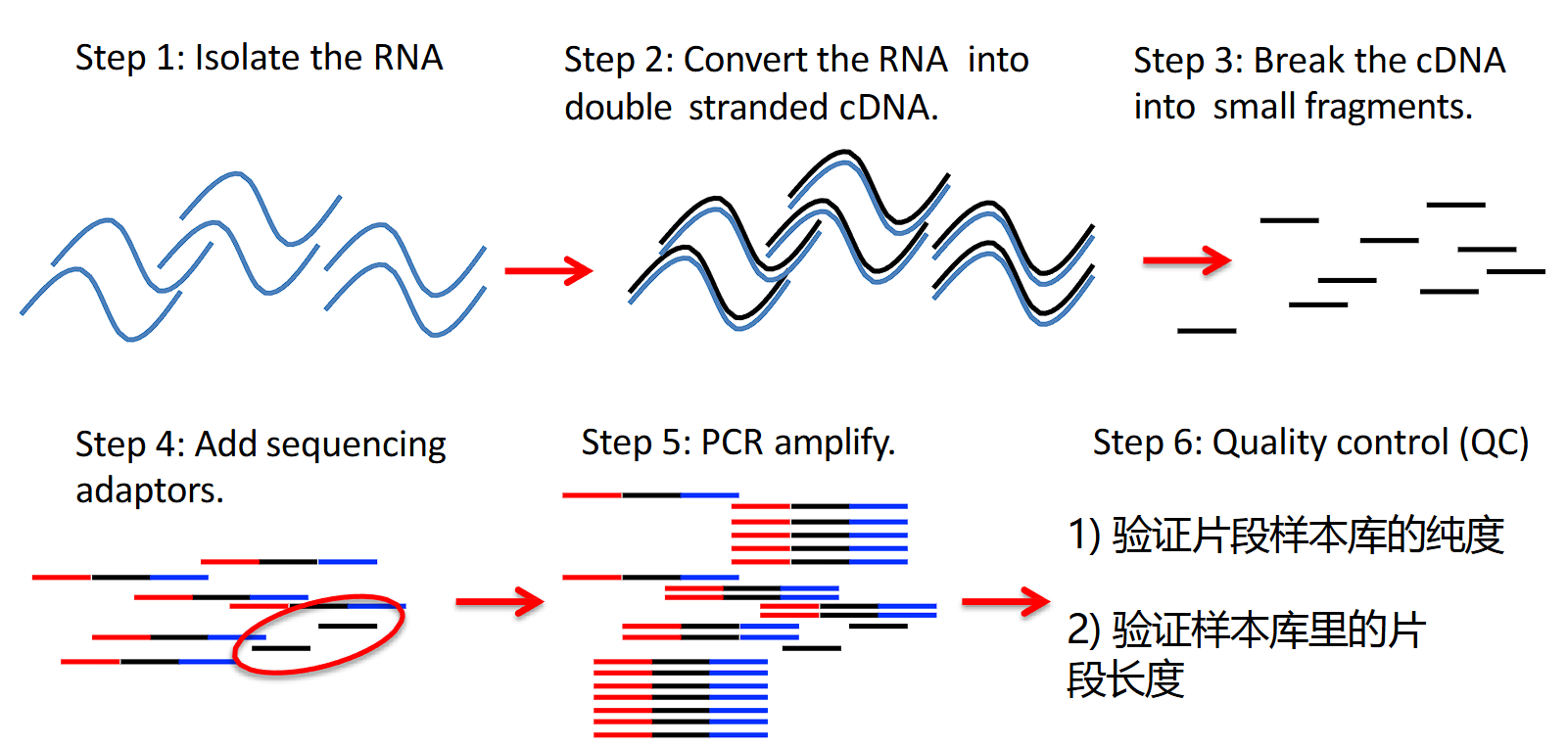
3 RNA测序与生物网络分析
RNA测序的目的是为了发现基因的表达差异
3.1 RNA测序
属于转录组学的一部分
转录组测序技术 RNA-seq
将各种RNA测序出来,快速获得某一物种特定器官或组织在某一状态下的几乎所有转录本
广义上指某一生理条件下,细胞内所有转录产物的集合
同一段RNA,经过剪切后可以形成不同功能的RNA,称为可变剪切异构体
由于RNA很不稳定,需要反转录为cDNA进行测序;这个cDNA和转录为RNA的DNA也是不同的,因为RNA会加工
圆圈内的配体没有匹配成功
表达量计算
不同数目的RNA进行测序,结果也会不同
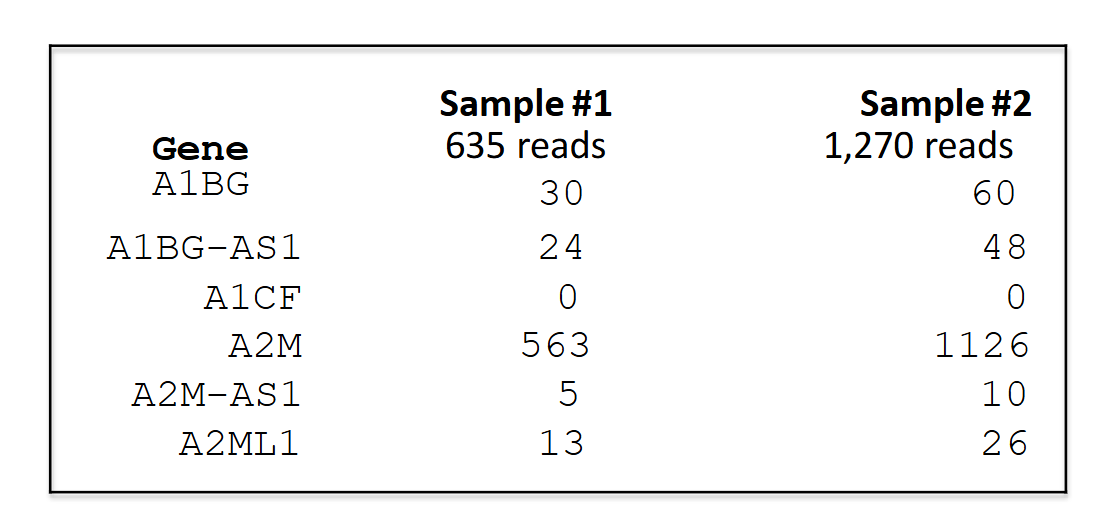
因为sample2的数目更多,测到的表达量也会更大
需要一些量对表达量进行归一化
- 基因长度不同
- 测序深度也不同
根据下面的假设:
- 在随机采样条件下,映射到特定转录本的读数计数与该转录本的丰度成正比
- 映射到转录本的读取数也与转录本的长度和总测序深度成正比例
有下面三种计算方式

- RPKM:适用于单端测序。基于每百万比对的单端读长的标准化表达值
- FPKM:适用于双端测序(将一对reads视为一个fragment)。对双端测序的改进版RPKM
- TPM :对RPKM/FPKM进行改进,保证样本内总和为固定值(即每百万转录本)。注重基因间的相对表达量
由于总和相同,能体现出比对上某个基因的reads比例,可以直接进行样本间比较
转录组测序项目整体思路
- 实验处理:寻找对照组和实验组
- 整体表达水平分析:整体表达水平分析;整体差异表达分析;差异基因聚类分析;差异基因功能富集
- 目标基因挖掘:目标基因挖掘,表达分析,功能分析,共表达分析,蛋白相互作用(根据已有研究)
- 后期验证:表达,定位,功能,其他水平(FPKM,TPM等)
样本的表达差异
对两组样本进行RNA测序后进行表达比对
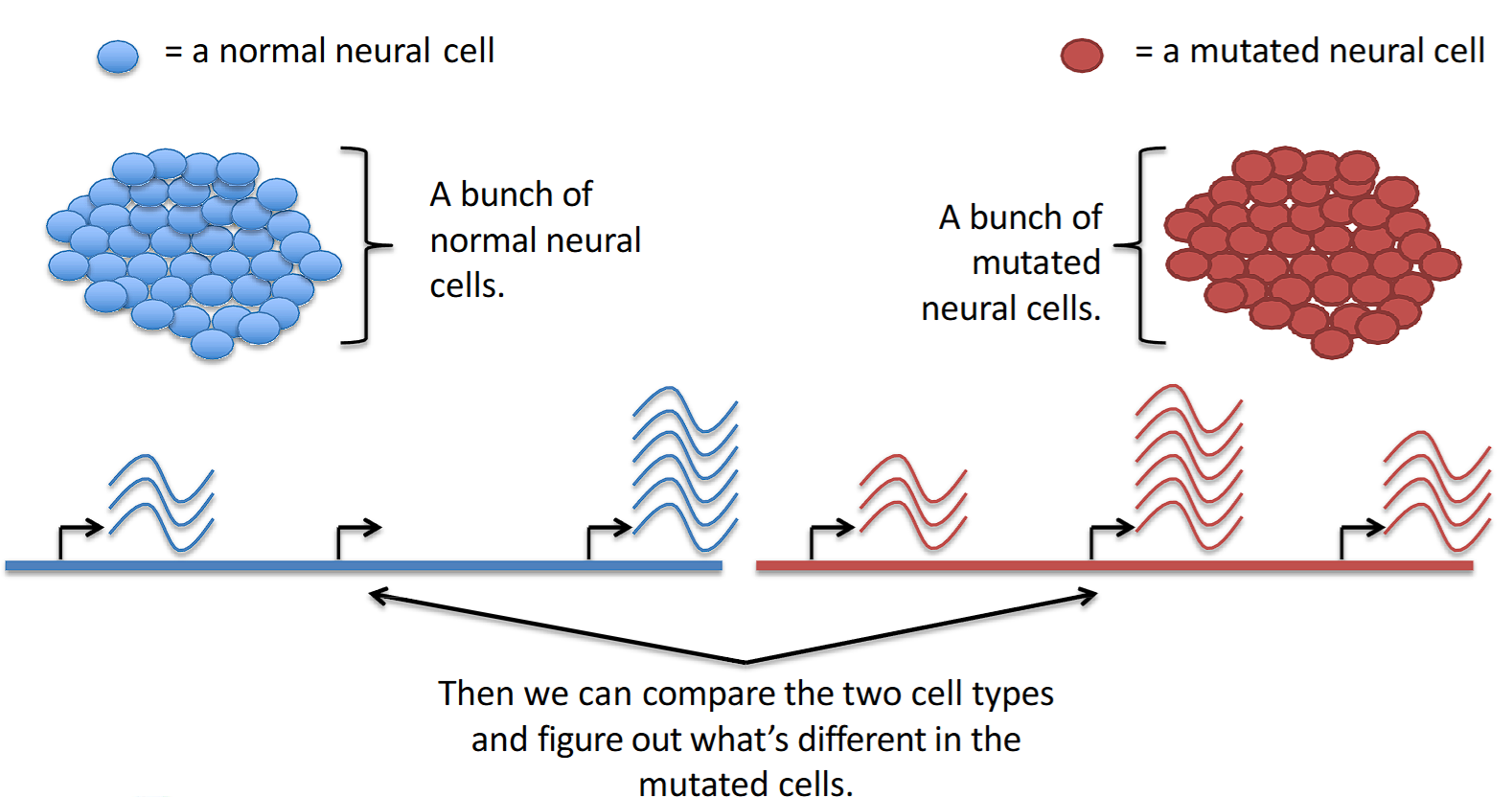
显著差异基因筛选
- Fold Change:两个样品中同一基因表达水平的变化倍数,>2为阈值
- q-value:多重假设检验矫正后的p-value,<.05为阈值
需要注意过滤低表达量的基因,否则有假阳性
3.2 WGCNA分析
背景
利用基因表达和调控方式来构建关系网络
- 基因间互相诱导(转录因子)或遏制(miRNA)另一个基因的表达
- 协调表达,相关的一组基因协调表达
以上两种会导致基因表达量间存在相关性;为了研究基因模块,而不是单一的基因,使用WGCNA
网络图
用图表示基因之间的关系,每个点表示一个基因,每条边表示调控关系
节点的度是这个节点的连线个数
无尺度网络
特点:大部分节点只和很少节点连接
分为随机网络和无标度网络
连通性
- 非权重网络:两个基因的相关性大于某个值认为其有关,且权重相同
- 权重网络:两个基因的相关性完全保留,用相关性表示权重
WGCNA原理
Weighted Gen Co-expression Network Analysis
最重要的是加权和共表达,筛除无效信息,缩小范围
WGCNA过程
数据清洗,去除离群值
- 进行聚类分析,看哪些样本不符合预期(剪枝离群值)
- 需要保证缺失值不能过多
构建共表达矩阵
- 构建相似度矩阵:cor
- 是中间变量,表示每对基因之间的相关性
- 用Pearson相关系数表示
- 选取分布两端5%的相关基因作为候选基因
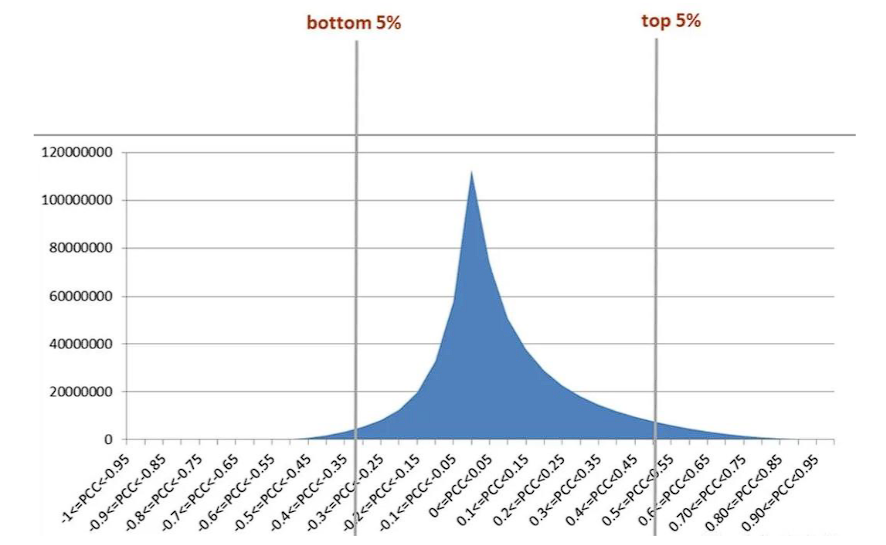
构建邻接矩阵:将相关性转化为连接强度(或权重):|cor|^β
利用相似度矩阵
使用Jackknife法(K折交叉验证):每次除去一个样本,重新计算相关系数,得到一组“Leave-One-Out”相关系数,用这些值评估整体相关性的稳定性或平均水平
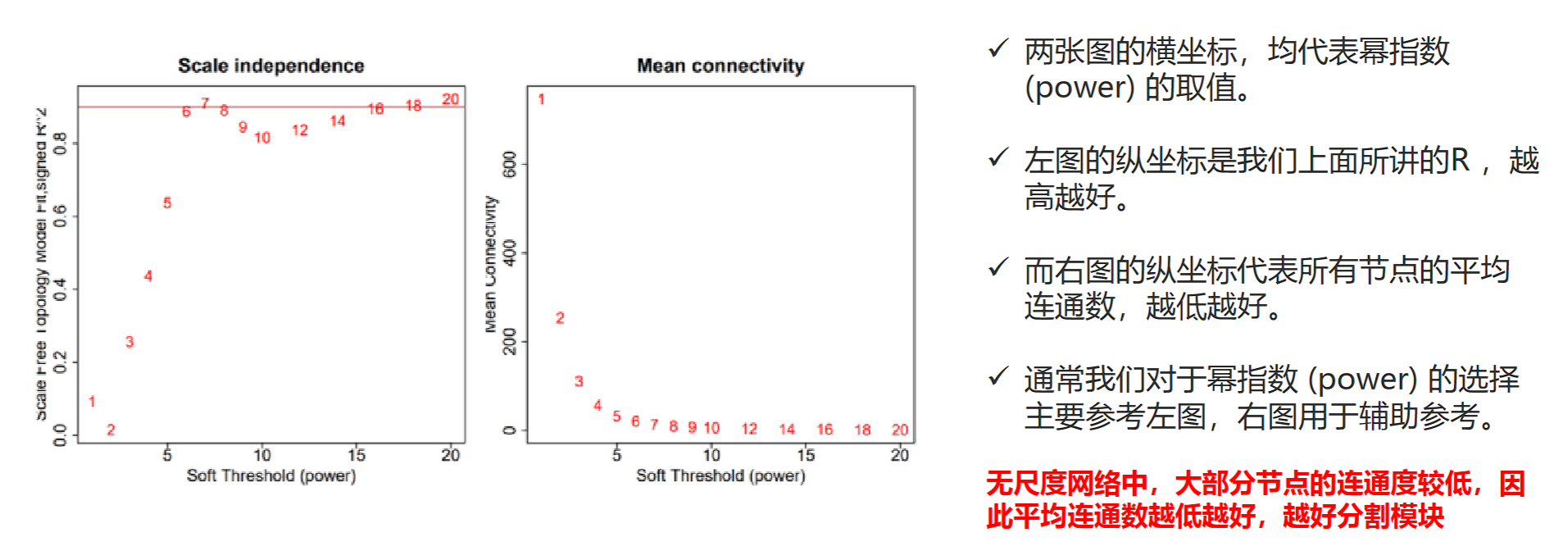
用soft-thresholding power β进行邻接系数计算
\[ a_{i,j} = |cor(x_i, x_j)|^\beta \]
一般用β=6或直接网格搜索寻找超参数如果要非加权图,那么用一个阈值过滤即可
下图中\(R\)是拟合优度;过大的power会稀释边缘连接强度,导致模块过小
构建拓扑重叠矩阵:TOM
从邻接矩阵计算而来
\[ TOM_{i,j} = \frac{\sum_u(a_{i,u}\cdot a_{j,u})+a_{i,j}}{\min(k_i,k_j)+1-a_{i,j}} \]
其中\(k\)表示节点的度邻接矩阵只考虑了直接相关性(边的强度),易受噪声干扰
TOM考虑了基因对之间是否共享相似的邻居,强调结构上的一致性
越接近1,表面有更多的共同邻居/直接相关性强
意义:
- 捕捉间接关联
- 增强网络鲁棒性
- 模块检测:按拓扑相似性聚类,识别基因模块
利用TOM进行聚类:将拓扑相似性转化为距离度量,再通过层次聚类和动态剪枝划分模块:1 - TOM
- 用1-TOM得到不相似性矩阵
- 距离越近越容易聚集
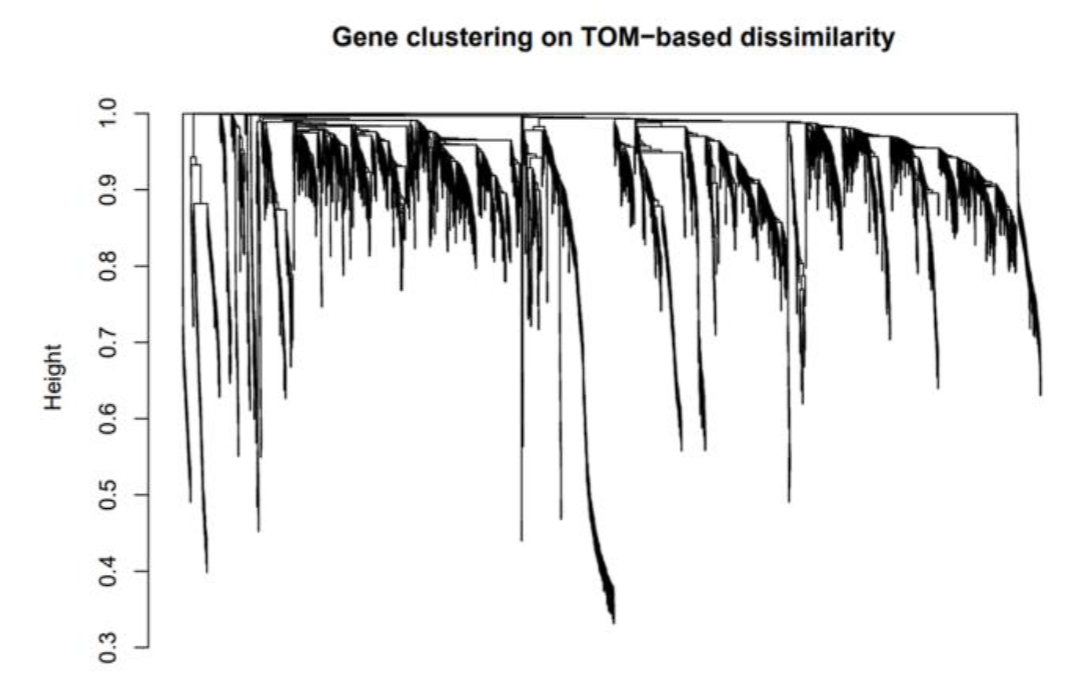
识别基因模块:连接十分紧密的基因集:hclust
- Dynamic Tree Cut算法,根据每一段树枝的结构复杂性、基因之间的紧密程度和树枝的独立性动态绝对是否剪断
- 对于每一个子树:计算内部一致性,与邻近分支的分离度;判断该分支是否独立/需要合并
- 自适应调整剪枝深度:小而密的分支,单独成模块;大而送的分支,再细分
- 可选PAM步骤精细化剪枝边界(尤其对边缘基因)
- Dynamic Tree Cut算法,根据每一段树枝的结构复杂性、基因之间的紧密程度和树枝的独立性动态绝对是否剪断
合并相似模块:手动将高度共表达的模块合并
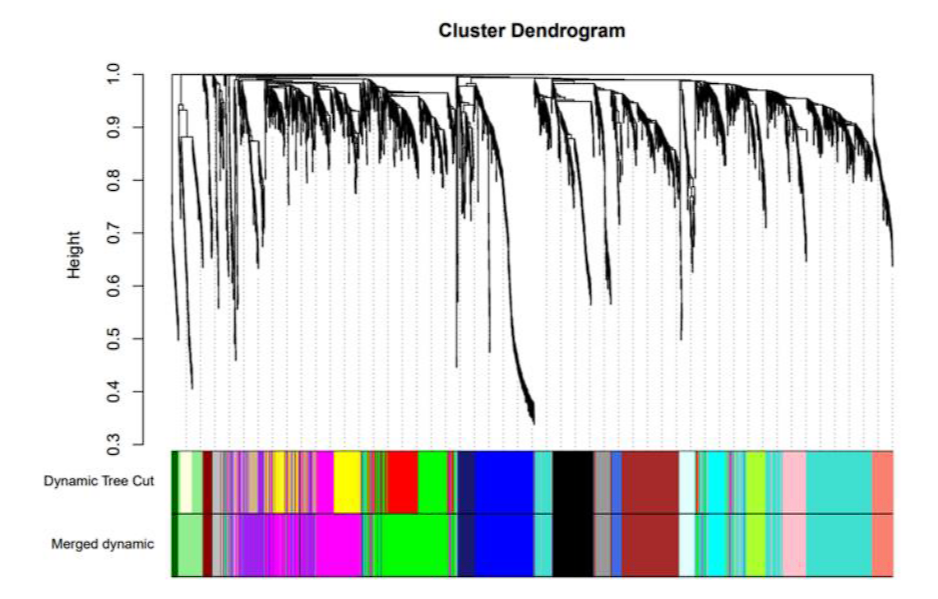
上面是第五步得到,下方是第六步合并后得到
模块功能验证:一个共表达模块反应一个真实的生物学信号
- 通常进行GO富集分析,见3.3节
模块与外部性状的关联
使用第六步得到的合并后的模块
将每个模块内的代表基因选出,作为模块特征基因
- 是一个代表表达趋势的虚拟基因
- 使用PCA进行降维
样本在各个模块的特征值可以和样本的性状进行相关分析
得到一个热图
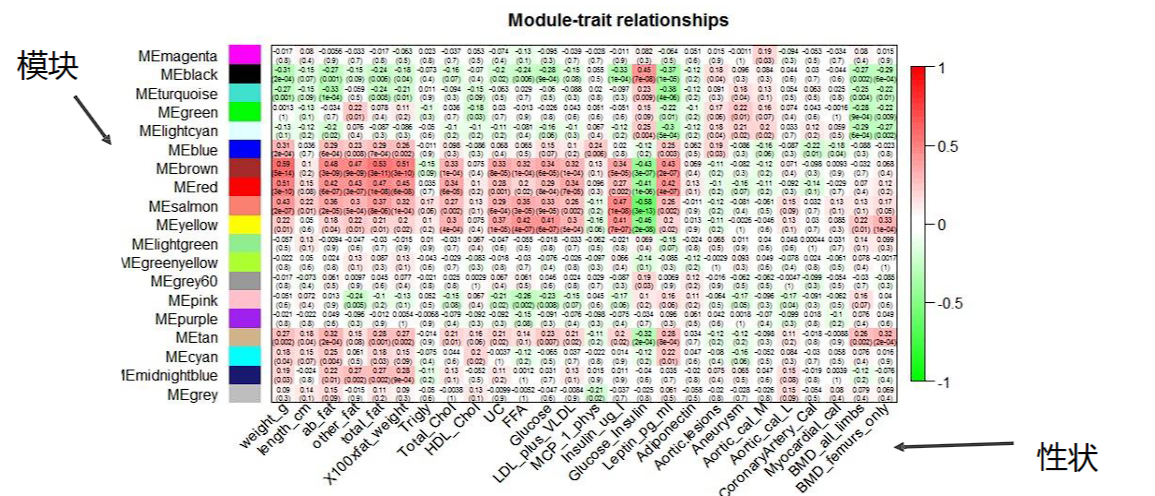
热图的每一个框内有两个数据
- 上方的数据是相关性大小,颜色深浅与绝对值有关,正向为红色,负向为绿色
- 下方的数据是相关系数的p-value
基因与外部性状的关联
注意:需要先选定一个模块
模块内存在很多基因,希望知道每个基因与模块对应的性状的相关性
定义一个新的变量GS(基因显著性),用散点图表示为
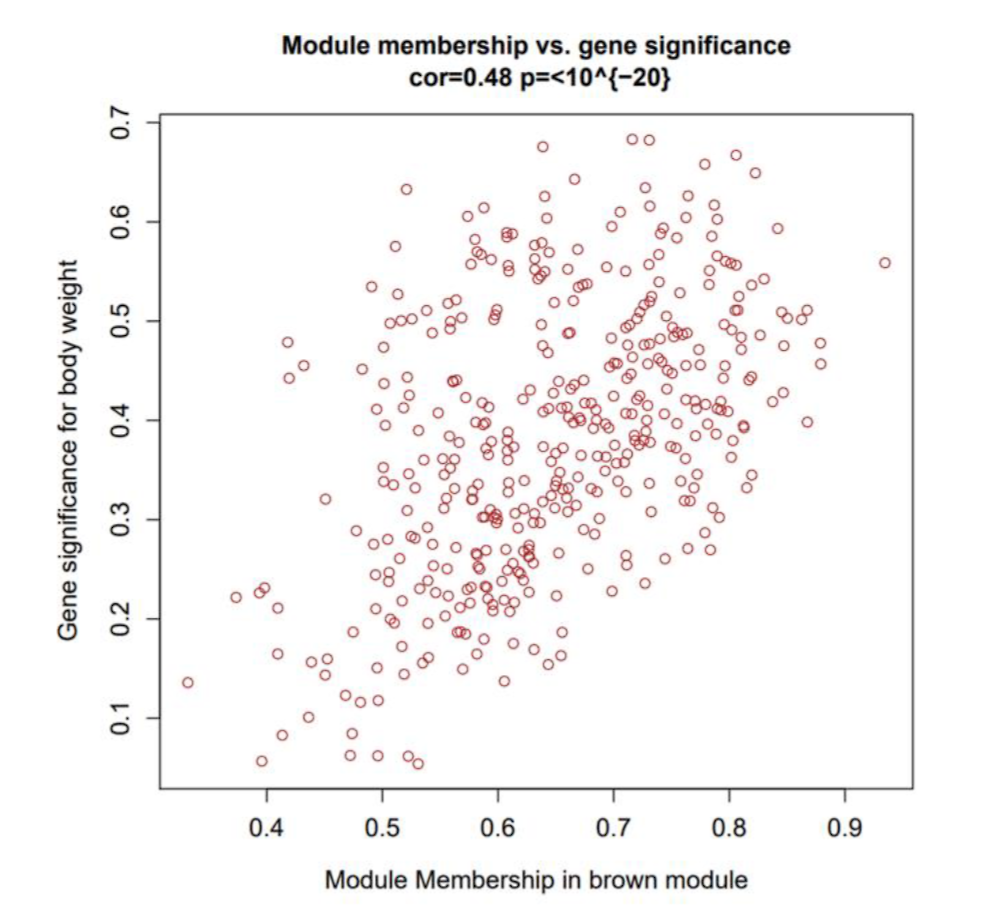
横坐标是基因在模块中的重要性(MM),纵坐标是基因在性状中的重要性(GS)
输出网络分析结果
后续分析
3.3 功能富集分析
3.3.1 数据注释
GO
Gene Ontology,基因本体论
- 三方面对基因及其产物予以注释:生物学过程,细胞组分,分子功能
- 统计注释到相应GO term上的基因数目/和其中的差异基因数目
KEGG
是一个数据库,注释过程使用到了KEGG Pathway通路图
- 利用图像展示代谢途径和各种途径的关系
- 直观反映基因表达差异对代谢通路的影响
- 将差异基因在通路上标注
- 全面了解代谢途径
3.3.2 数据挖掘
使用基因富集分析,找出表达水平有差异的基因集
计算过程
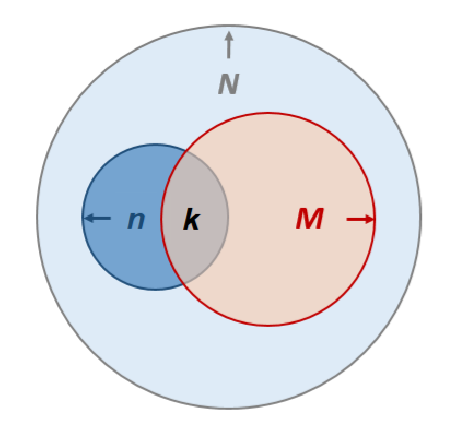
符号:N表示全体基因数据;M表示GO中的基因;n表示数据库中有关基因;k表示M和n的交集
背景命中率为\(\frac{n}{N}\),GO列表中的命中率为\(\frac{k}{M}\),用两个值做假设检验就知道GO的结果的显著性
更准确的来说,在富集分析中,想要知道“在特定的列表中,一个特定的基因是否比随机选择的情况下出现得多”
类比于选次品的问题,零假设是出现的概率没有更大,那么就符合超几何分布
\[
P = 1- \sum^{k-1}_{i=0}\frac{\binom{M}{i} \binom{N-M}{n-i}}{\binom{N}{n}}
\]
记为\(X\sim \mathcal{H}(n,M,N)\),当\(P<.05\)时认为零假设不成立
当然在多重检验下需要修正概率,有两种方法,第二种常用,选其一
Bonferroni Correction,For Family Wise Error rate
使用检验的次数归一化p-value,例如检验1000次,则阈值设为\(0.05 / 1000\)
Benjamin-Hochberg Correction, For FDR
每次检验的p按照序号设置阈值,如序列\(p_1,p_2,...,p_s\),其中\(p_i \leq \alpha \cdot\frac{i}{m}\),\(m\)是总的检验次数
3.3.3 Metascape
一站式功能富集工具,采用CAME通用分析工作流程
- ID转换,将用户输入的基因表示符自动转换为目标物种的entrez gene IDs
- 注释,为基因添加描述,功能和蛋白质分类等
- 归类,获取感兴趣的基因集(存在多个,产生多个富集通路)
- 富集
分析结果
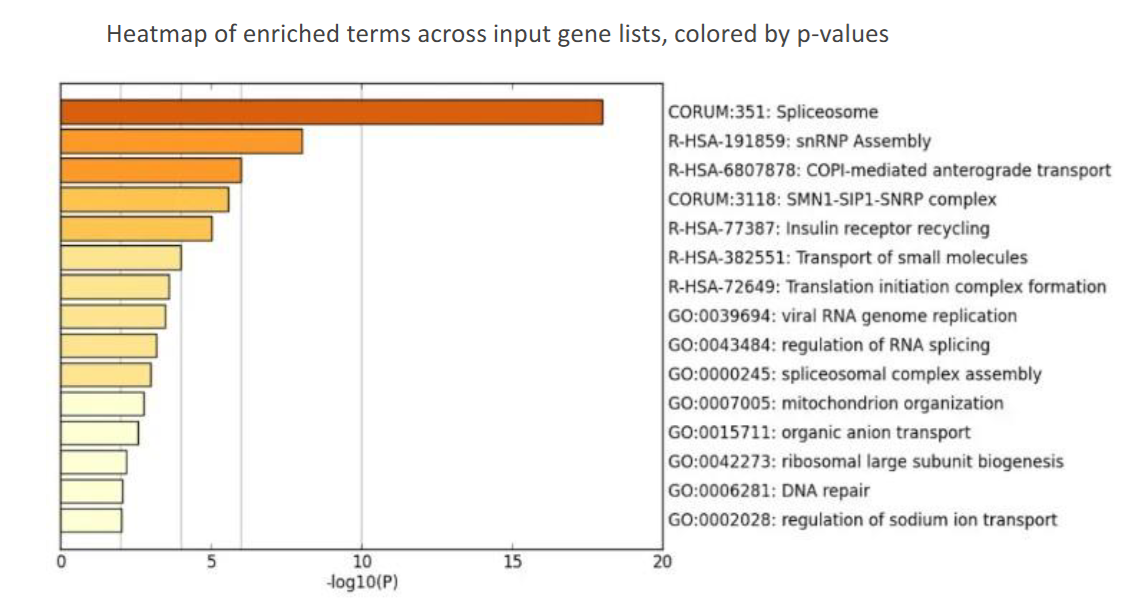
横坐标是\(-\lg p\),越小的p会越大;纵向是不同的富集通路,约上面的富集越显著
3.4 然后需要做什么
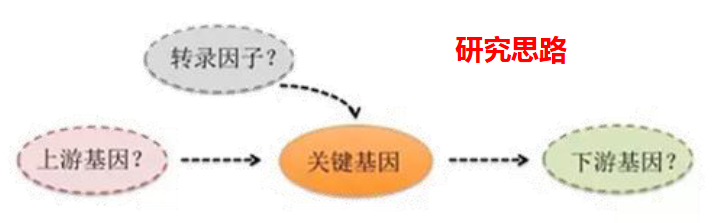
在找到关键基因后,还有三个方向可以干