2 DNA测序与突变
2.1 基因测序技术简介
组学:是研究生物系统中各类分子整体集合的学科,以-omics结尾
- 基因组学、转录组学、蛋白质组学、代谢组学(糖组学、脂类组学)
一代测序:多分子,单克隆;桑格测序;高精度检测序列,用于亲子鉴定、法医鉴定等
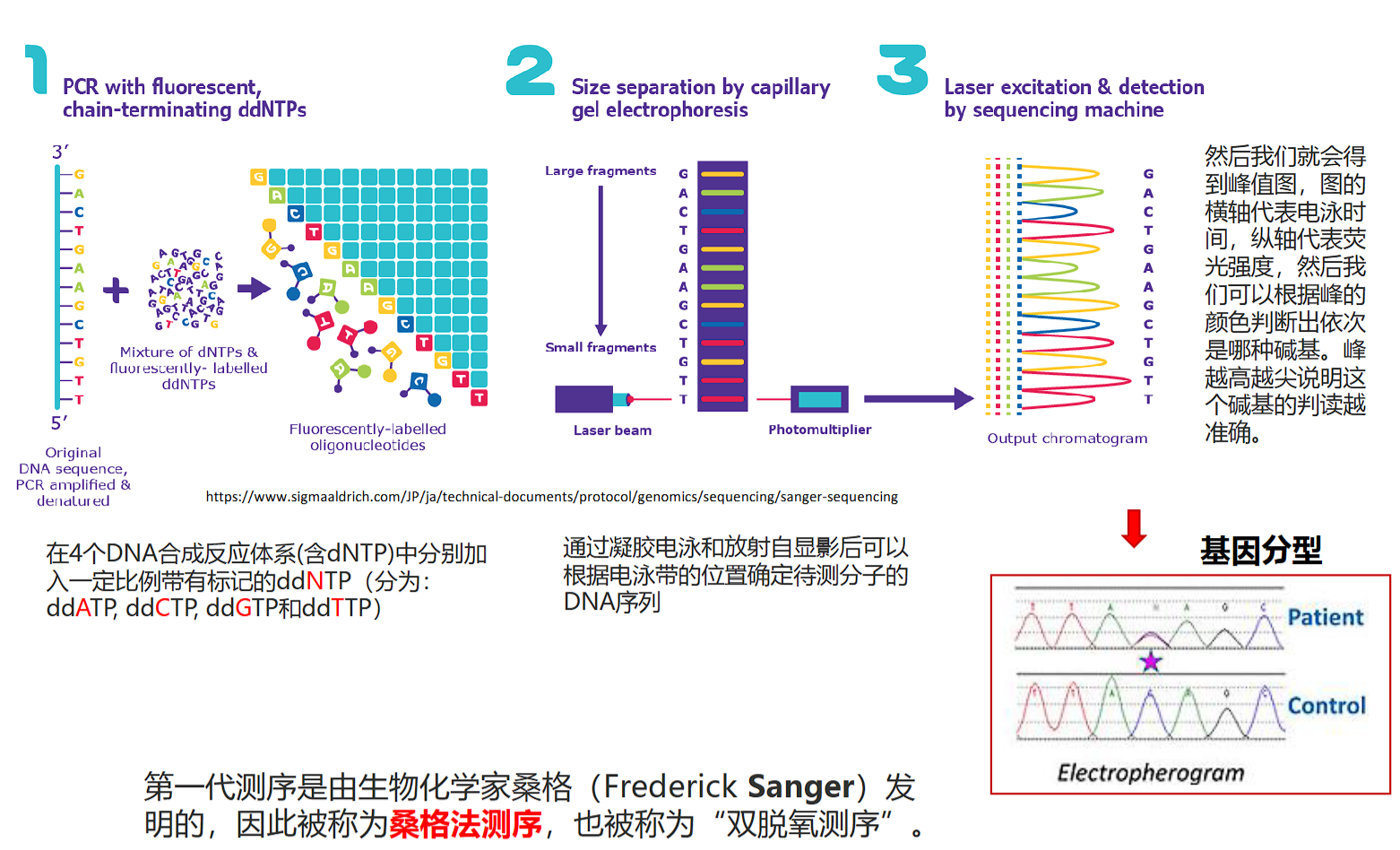
二代测序:通量更高,速度更快,成本更低
2.2 基因突变
2.2.1 基因突变
同一基因库之间不同个体存在DNA水平差异
- 遗传
- 变异
物种形成和生物进化的基础
遗传变异来源
- 父母遗传
- 新发突变
- 体细胞突变
几种对比

多态性指的是性状不同
健康人基因组存在300万左右的变异
单位点变异
- 错义突变(missense mutation):碱基替换后对应的氨基酸变了
- 无义突变(nonsense mutation):碱基改变后使的密码子变为终止密码子
- 同义突变(synonymous/silent mutation):碱基替换后氨基酸没变
单核苷酸多态性(SNP):平均每300个碱基对就存在一个;SNP的多态性决定了个体差异
表型 = 基因型 + 环境
indels: insertion or deletion,碱基对的变化
2.2.2 基因测序和突变识别
包括全外显子组测序和全基因组测序
基因变异识别流程
- 建库测序
- 质检,数据清洗
- 读数映射
- 变异识别
- 有害突变识别和个性化诊疗
质检
得到的结果如下

最后一行是质量分数对应的ASCII码对应的字符,其中质量分数(Phred quality score)的计算如下
\[
Q = -10\cdot \lg p
\]
\(p\)表示测序仪碱基判断错误的概率
当\(Q>20\)时,认为结果有效;当一个序列\(Q<20\)的比例达到20%,认为这个序列读错了,要丢弃
例:当一条read的得分不高时
- 不直接使用,因为质量很差
- 若认为是全外显子组,则可能产生假阳性突变识别的结果,影响后续的变异分析
- 当read质量高且覆盖深度不足时,一个read也可能预警潜在突变
变异识别
基因序列比对后进行识别
例:TP53基因某位点发生非同义突变,已知是非保守位点
这个位置值得关注,因为TP53是典型的肿瘤抑制基因
如果是保守区域,那更值得关注。保守区域暗示重要功能,突变更可能破坏关键结构
一句话报告:
该突变为TP53基因非同义变异,位于潜在保守功能区域,建议进行家系分析和功能验证
临床使用
读数深度很重要,读了几条就是几,一般要大于三
2.3 全基因组关联分析 GWAS
Genome-Wide Association Study,分析比表型组和基因组的关联
使用全基因组阵列收集数据,以便在多个个体中找出基因组中具有/不具有共同特征的共同变异
用高通量基因组技术快速扫描大量受试者的整个基因组,寻找与疾病关联的相关变异;与疾病有关的变异在病例组中的发现频率更大,做统计检验
很显然这需要进行假设检验,这就要看生医统的知识了
整个检验就流程如下
- 基因型数据准备,表型数据准备
- 假设检验(统计模型选择)
- p值计算
- 绘图,结果解读
假设阳性概率为\(p\),\(n\)个人,那么假阳性概率为
\[
1-(1-p)^n
\]
为了平衡这个概率,需要用人数归一化
\[
\frac{1-(1-p)^n}{n} = \frac{np}{n} = p
\]
那么,对应的显著性水平就从\(p<.05\)变为\(p<\frac{0.05}{n}\)
当数据水平是百万个SNP时,\(p<5\times10^{-8}\)
2.4 孟德尔随机化
2.4.1 疾病风险评估
GWAS的p-value确定检验目标是否有统计学意义,但没有解释影响效果大小,即不能解释GWAS的结果
因此,需要一个方法能够评估,存在遗传变异的情况下,患上疾病的风险是多大
风险评估
用人群中发病率表示发病的风险,但这种方法对于个体差异没什么意义
相对风险
想要知道有某种基因的风险相对没有的人的发病风险,即相对风险,就是两种风险的比值(11% / 9% = 1.2)
但是上面的方法还有一个问题,对于罕见病,人群中的样本数量不足;做常见病倒是可以
病例对照研究
在疾病对照研究中,预先确定500个样本的病例组和500个样本的对照组,不能说明发病率为0.5
但是,可以说明患病几率(Odds),Odds = disease : no disease = 1:1
对于所有样本中的基因型比例,可以计算出Odds值
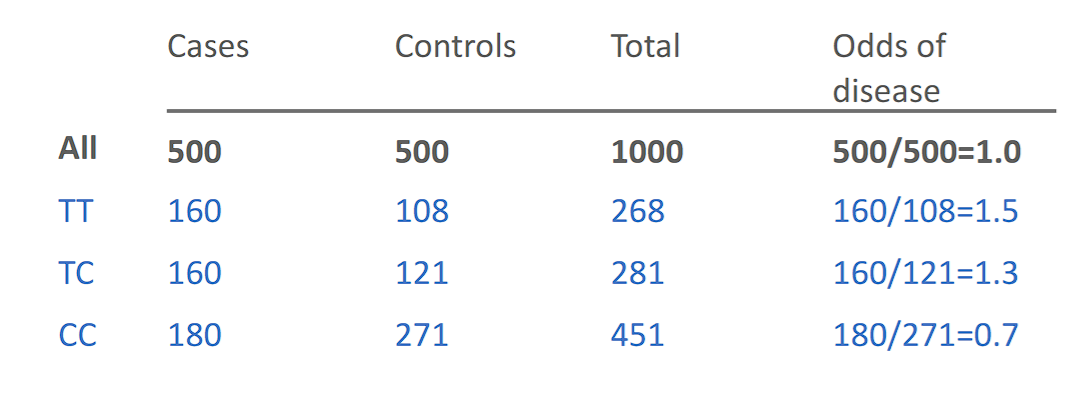
Odds值较大的,说明这种基因型发病概率高
同样可以做相对风险,比值比 Odds ratio = Odds1 : Odds2,例如 TT : CC = 1.5 : 0.7 = 2.1
2.4.2 孟德尔随机化
一种基于遗传变异的统计方法,评估病因推断的数据分析技巧;MR的目的是明确因果关系
临床上的金标准是RCT(Randomized Control Trial),但是开销大,伦理可能有问题,此时可以用MR
MR的几个关键概念
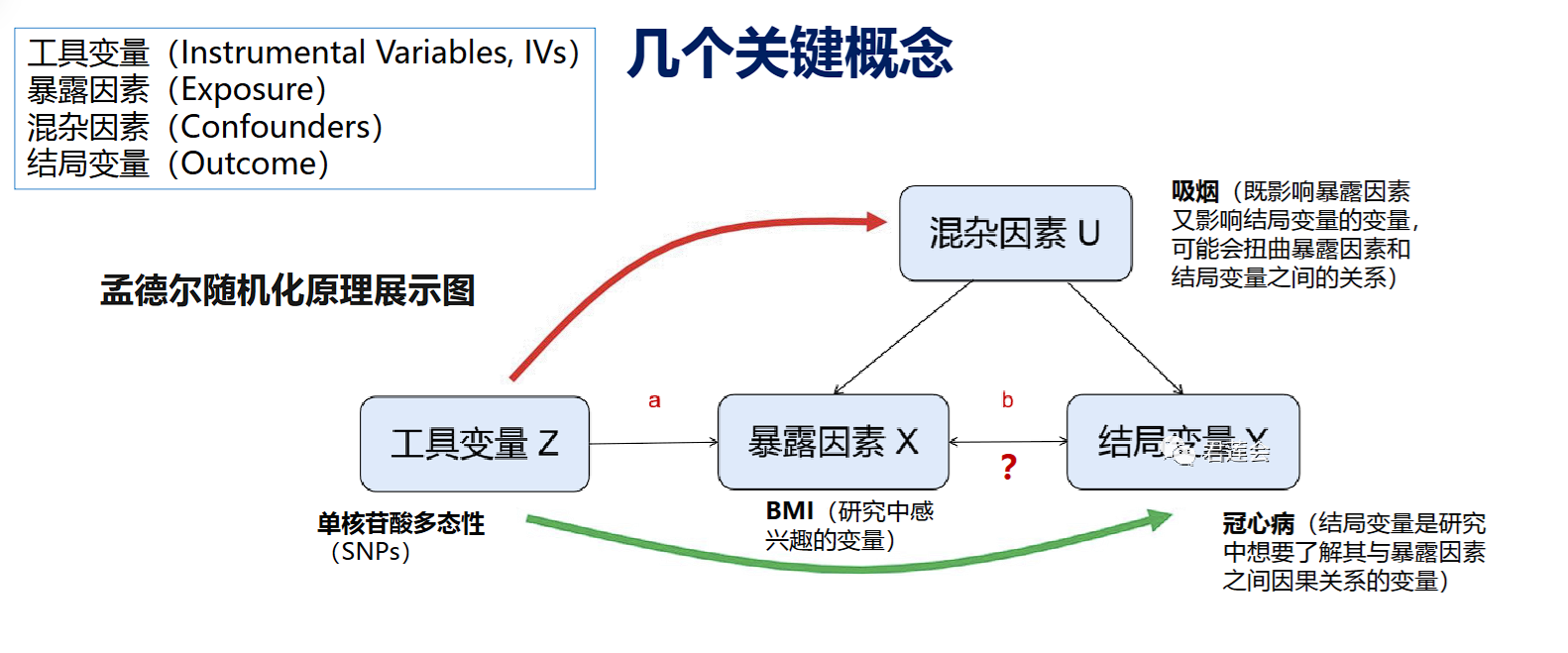
常规的RCT难以进行的原因就是混杂因素导致暴露因素和结局变量因果倒置,或者是伦理问题
为了得到有效的结果,必须满足三个核心假设
- 遗传变异与暴露因素强相关(关联性)
- 遗传变异不能与任何可能的混淆因素相关(独立性)(图像红线不通)
- 遗传变异不能与结果直接相关(排他性)(图中绿线不通)
两样本MR流程
- 暴露因素的GWAS:对暴露因素进行GWAS,识别出与暴露因素相关的显著SNPs(p值小)。
- 结局变量的GWAS:对结局变量进行GWAS,检查暴露因素相关的SNPs在结局变量中的关联;去除与结局变量显著相关的SNPs,确保工具变量与结局变量没有直接关联。
- 筛选工具变量:保留那些与暴露因素显著相关、与结局变量无显著直接关联的SNPs作为工具变量。
- 敏感性分析(可选):进行如MR-Egger回归等方法,确认工具变量的排他性条件是否满足。
RCT通过随机分组对抗混杂因素的干扰,用于分组的标准应当满足
- 决定治疗方法
- 与结局没有直接关联
- 独立于混杂因素
而MR借助核心假设确定工具变量,绕开了混杂因素的干扰
MR根据孟德尔第一、二定律(分离定律和自由组合定律),利用基因型作为工具变量
二者对比如下
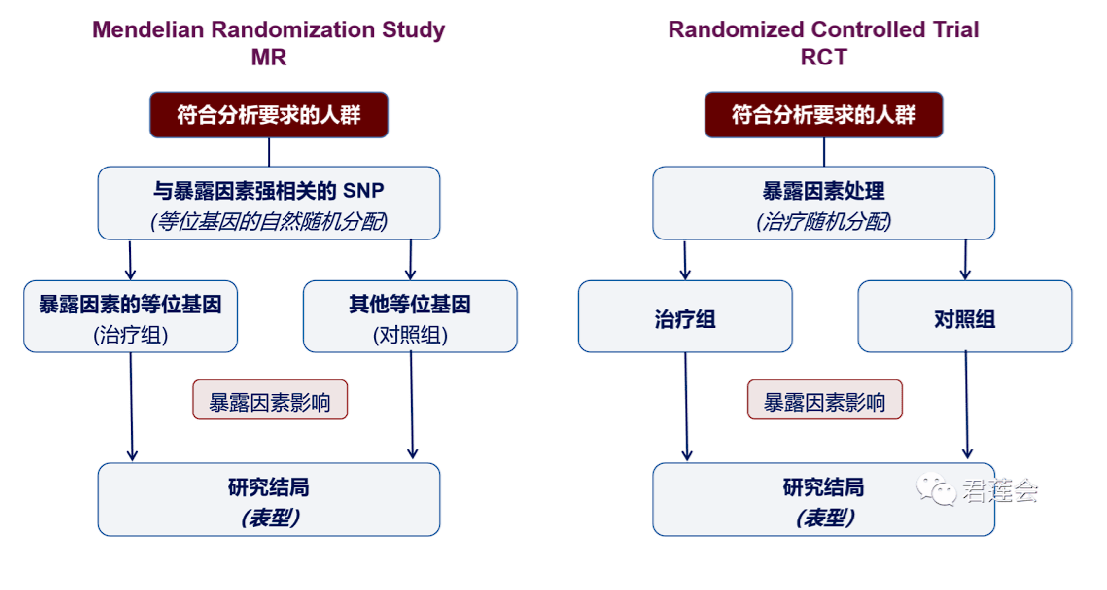
MR的本质和原理
- 疾病发展是多基因和多因素共同作用的结果
- 目前GWAS已经有足够的数据,发现了百万的遗传变异与疾病相关联
- 本质是通过遗传数据来评估可改变的非遗传暴露因素所造成的因果效应
分析流程
- 读取GWAS数据,选择合适的工具;一般设置 \(p<5\times 10^{-8}\)
- 读取结局变量的GWAS数据,提取变异频率大于1%的单核苷酸变异
- 数据预处理
- MR分析SNPs与结局的关系:调包
- 结果后处理
2.5 突变功能注释
对应于2.2.2节的有害突变识别和个性化诊疗
假设:如果突变残基和野生型残基之间的替换分数为正,则为中性突变(很容易发生的损害小);反之,是有害的(发生很少的损害大)
2.5.1 SIFT
给定一段蛋白质序列和核酸的变异:
- 首先搜索与待测序列相似的序列,用来提供氨基酸替换的背景;即把同源的蛋白寻找出来
- 选择1的结果中与查询序列功能最相近的序列,进行多序列比对,确定氨基酸的位置和保守性
- 根据2的结果,计算所有的替换是正常的可能性,如果<.05,就是有害的;如果>=.05,就是中性的,不影响表型和功能
但是SIFT的效果并不好,原因是只靠保守性假设不靠谱
2.5.2 PolyPhen
依靠序列比对和蛋白质三维结构进行判断,主要假设
- 保守区域的变异影响更大(保守性)
- 发生在蛋白质活跃位点,交互位点或蛋白质的稳定性的变异影响大(功能性)
- 蛋白质的三维结构改变影响很大(结构变化对功能的影响)
步骤和SIFT有点像
- 多序列比对,计算序列的特征值
- 获取蛋白质的三维结构,或者预测结构
- 在一些特定位点上计算变异和分类
- 使用基于经验的方法进行损害预测
难点在于三维结构的获取;最终打分区间为0-1,分越高越有害
2.5.3 CADD
整合多种功能预测工具的数据,利用支持向量机(SVM)将复杂的注释信息浓缩为单一评分
最终用对数打分,有害前1%打分为20,10%为10,越大越有害